Proteomics <br> Proteomics is the science of identifying and quantifying proteins in cells, tissues, or organisms to understand biological changes and disease states, and to develop biomarkers for disease and therapeutic targets.
If each modified protein of a basic protein is counted as a protein, the mammalian cells contain up to 30,000 to 40,000 proteins. When calculating each modified protein, the amount of protein far exceeds 100,000.
The abundance or concentration of different proteins within a cell may vary by orders of magnitude.
Any change in the cellular system, such as aging, illness, or medication, can result in a change in the relative abundance or expression of one or more proteins.
Proteomics aims to identify proteins within the cellular system and to identify and monitor changes in proteins, such as modifications to proteins (post-translational modifications) or changes in the relative concentration of proteins.
If an event increases protein abundance, then we say that the event upregulates protein expression. If an event causes a decrease in protein abundance, then we say that the event downregulates protein expression.
Protein abundance may change significantly when affected by cellular changes, and changes in protein abundance are signals of event changes.
Post-translational modification (PTM) refers to the chemical modification of a protein after translation.
PTM includes addition of an oligosaccharide chain (glycosylation), N-terminal modification of an alkyl group-containing protein such as acetate (acetylation, etc.), addition of a phosphate group on serine, threonine or tyrosine (phosphorylation) Similar activities.
PTM activity, such as phosphorylation, is largely temporary and is used for the transfer of intracellular and intercellular information.
PTM activity, such as glycosylation, is a structural change that typically affects protein folding, interaction with other molecules, and interaction with cell walls.
All of these represent dynamic changes that occur within living cells.
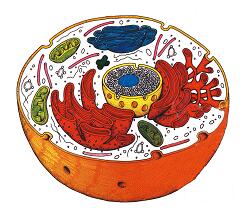
A proteome indicates all proteins in a cell or in any study sample.
A complete cellular proteome consists of 40,000 individual proteins with up to 10 to 20 different modifications, and abundance varies by order of magnitude.
The goal of proteomics is clearly huge, requiring the most powerful separation and analysis techniques.
Electrophoresis A
In order to achieve identification and/or quantification of proteins, individual proteins must be separated from other proteins as much as possible.
For many years, gel electrophoresis has been the primary means of isolating intact proteins.
Two-dimensional gel electrophoresis (2DGE) achieves protein separation based on the isoelectric point (first direction) and molecular weight (second direction) of the protein (see Figure 48).
The 2DGE of the intact cell proteome is quite complex, and it is difficult to separate proteins from each other while separating as many as 2000 to 3000 proteins, or it is difficult to see on gels due to the low abundance of certain proteins. 2DGE is still a powerful protein separation tool, a well-developed technology with many experienced users, capable of high resolution and quantitative protein.
At present, the limitation of 2DGE is that it is not only time-consuming and laborious, but mainly measures the protein with higher abundance, and it is difficult to measure important proteins such as membrane-bound protein.
Figure 48. Two-dimensional gel electrophoresis can separate up to 2000 to 3000 individual proteins.
Each spot represents one or more proteins.
Lateral separation is caused by the isoelectric point and is related to the charge.
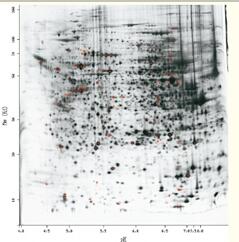
Longitudinal separation is caused by molecular weight (SDSPAGE).
The smaller molecular weight protein moves to the bottom of the gel, while the larger molecular weight protein remains on top of the gel.
Chromatography <br> Chromatography has evolved into a powerful separation technology that separates large amounts of proteins and peptides.
But any single chromatographic technique can still only isolate a small amount of protein.
Therefore, the combined use of multiple chromatographic techniques has become a common method for protein separation in proteomic analysis.
2D Chromatography <br> Based on a long-term protein purification model, John Yates and colleagues developed a proteomic analysis technique called MudPIT.
In this technique, a proteolytic enzyme such as trypsin is first used to hydrolyze proteins in the proteome into polypeptides having a relatively small molecular weight.
These peptides were subsequently separated by ion exchange chromatography by increasing the salt concentration stepwise.
At each step, the salt concentration was slightly increased, and the peptide with weaker binding ability was eluted to the reverse phase magnetic beads. The mobile phase is an acetonitrile gradient eluate with hydrophobic elution of the polypeptide.
In the MudPIT method, ion exchange beads and reverse phase beads are sequentially loaded into a capillary column, and the eluate flowing from the reverse phase portion directly enters an electrospray mass spectrometer (Fig. 49).
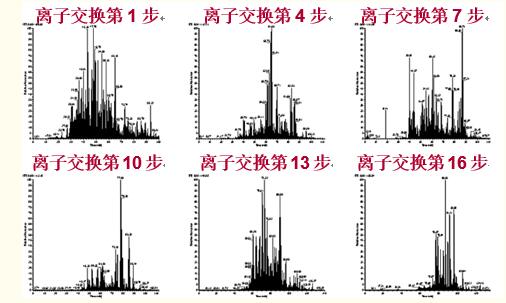
This process is repeated multiple times to enable important separation of the proteolytic product peptides (Figure 50).
The advantage of two-dimensional chromatography for the separation of protease hydrolysate polypeptides is the decomposition of proteins into peptides, allowing protein separation and identification not possible with gel electrophoresis, such as some hydrophobic membrane-bound proteins and low-abundance proteins.
The disadvantage is that information about the PTM is usually lost in the MudPIT method.
In addition, the number of peptides formed is much larger than that of proteins, and on average, each protein is hydrolyzed to obtain 20 to 50 peptides, and must be isolated.
Figure 49. Multidimensional Protein Identification Technique (MudPIT) consists of a preliminary separation of the entire proteome protease hydrolysate by ion exchange chromatography and reversed phase separation of the polypeptide in the fragment by ion exchange.
The ion exchange magnetic beads and the reverse phase magnetic beads were loaded into a capillary column, and the eluate flowing from the reverse phase portion directly entered the electrospray mass spectrometer.

Figure 50. In the multidimensional protein identification technique (MudPIT), a gradual increase in salt concentration elutes the polypeptide to the reverse phase of the column, and the acetonitrile gradient eluate separates the polypeptide according to hydrophobicity.
PTM information is usually lost in the MudPIT method.
In addition, the number of peptides formed is much larger than that of proteins, and on average, each protein is hydrolyzed to obtain 20 to 50 peptides, and must be isolated.
A more advanced approach is to use affinity chromatography, immobilized metal affinity chromatography and lecithin affinity chromatography to further separate subfragments of the polypeptide before or after ion exchange separation.
Protein Identification <br> Proteins separated by gel electrophoresis are usually hydrolyzed by proteases (in most cases, trypsin), and the resulting polypeptides are analyzed by MALDI mass spectrometry and identified by means of a protein database.
Peptides separated by MudPIT and related methods were introduced into an electrospray mass spectrometer and identified by mass spectrometry and tandem mass spectrometry using a protein database.
When several peptides of a certain protein are identified, the protein can be identified accordingly.
Isolation Fence,Zinc Steel Traffic Fence,Traffic Fence,Fences Road Isolation
Xuzhou Guifeng Metal Technology Co., Ltd , https://www.guifengmetal.com